Meet MaxToki: The AI That Predicts How Your Cells Age — and What to Do About It
Most foundation models in biology have a fundamental blind spot: they see cells as frozen snapshots. Give a model a single-cell transcriptome — a readout of which genes are active in a cell at a given moment — and it can tell you a lot about what that cell is doing right now. What it can’t tell you is where that cell is headed.
That limitation matters enormously when studying aging. Age-related diseases like heart disease, Alzheimer’s dementia, and pulmonary fibrosis don’t happen overnight. They unfold across decades, driven by slow, progressive shifts in gene network states. To understand and eventually reverse these trajectories, you need a model that thinks in time — not just in snapshots.
That’s exactly what MaxToki is designed to do.
What MaxToki Is, Under the Hood
The team involved in this research includes researchers from institutions like the Gladstone Institute of Cardiovascular Disease, the Gladstone Institute of Data Science and Biotechnology, and the Gladstone Institute of Neurological Disease, all alongside the University of California San Francisco’s Division of Cardiology, Biological and Medical Informatics Graduate Program, Department of Pathology, Department of Neurology and Bakar Aging Research Institute, Department of Pediatrics and Cardiovascular Research Institute, and Institute for Human Genetics. Also contributing were the University of California Berkeley’s Department of Molecular and Cell Biology and NVIDIA along with the Institute of Cardiovascular Regeneration and Centre for Molecular Medicine at Goethe University Frankfurt, the German Center for Cardiovascular Research, the Cardiopulmonary Institute, and the Clinic for Cardiology at University Hospital Frankfurt from Germany, and the Center for iPS Cell Research and Application at Kyoto University. MaxToki is a transformer decoder model — the same architectural family behind large language models — but trained on single-cell RNA sequencing data. The model comes in two parameter sizes: 217 million and 1 billion parameters.
The key representational choice is the rank value encoding. Rather than feeding raw transcript counts into the model, each cell’s transcriptome is represented as a ranked list of genes, ordered by their relative expression within that cell after scaling by expression across the entire pretraining corpus. This nonparametric approach deprioritizes ubiquitously expressed housekeeping genes and amplifies genes like transcription factors that have high dynamic range across distinct cell states — even when lowly expressed in absolute terms. It’s also more robust against technical batch effects, since relative rankings within a cell are more stable than absolute count values.
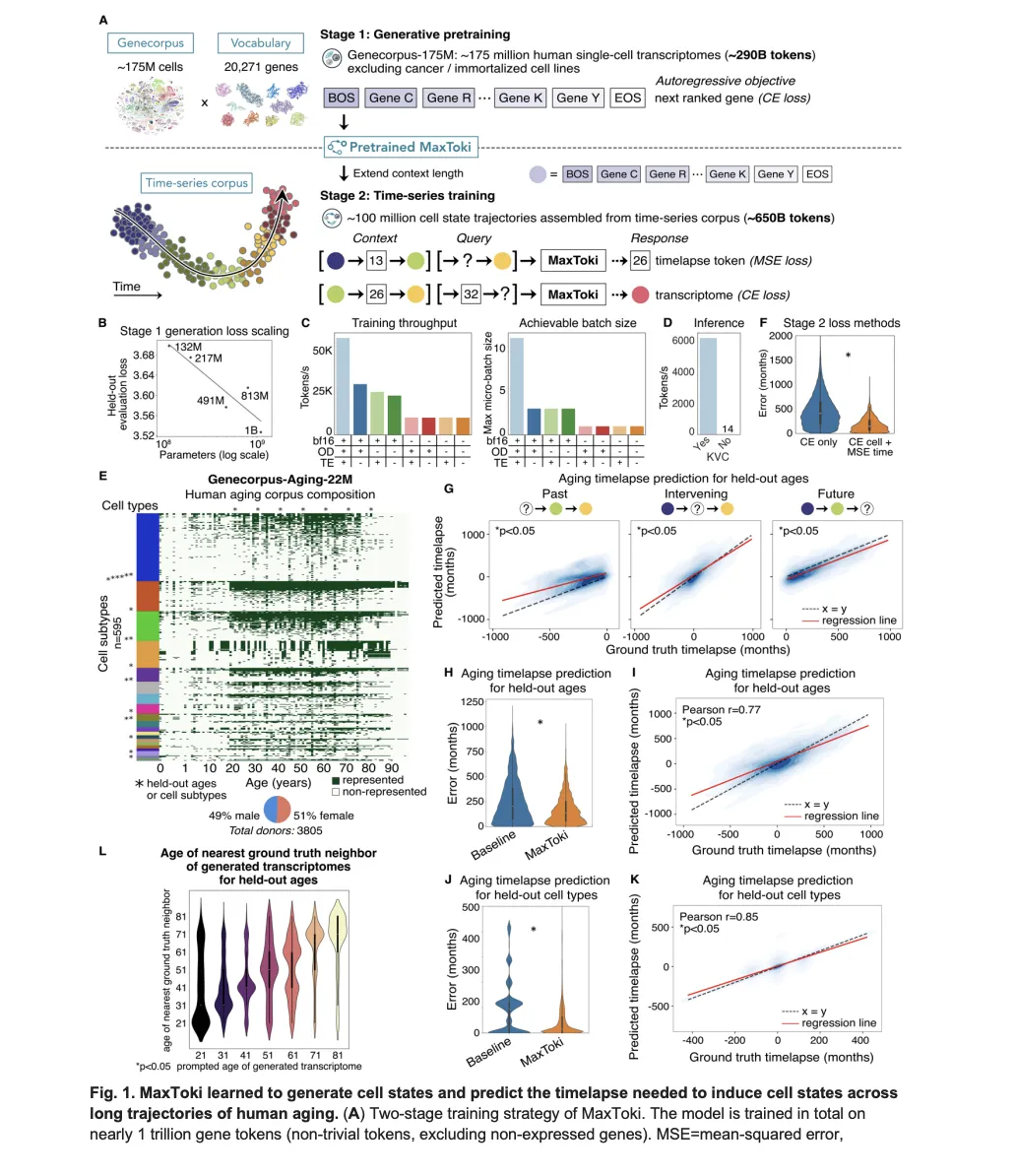
Training happened in two stages. Stage 1 used Genecorpus-175M — approximately 175 million single-cell transcriptomes from publicly available data across a broad range of human tissues in health and disease, covering 10,795 datasets, generating approximately 290 billion tokens. Malignant cells and immortalized cell lines were excluded because their gain-of-function mutations would confound what the model learns about normal gene network dynamics, and no single tissue was permitted to compose more than 25% of the corpus. The model was trained with an autoregressive objective: given the preceding genes in the rank value encoding, predict the next ranked gene — conceptually identical to how language models predict the next token in a sentence.
A key technical finding from Stage 1 is that model performance on the generative objective scaled as a power law with the number of parameters. This motivated the choice to fully pretrain exactly two variants — the 217M and 1B — rather than exploring the full spectrum, balancing performance against compute budget constraints.
Stage 2 extended the context length from 4,096 to 16,384 tokens using RoPE (Rotary Positional Embeddings) scaling — a technique that interpolates more tokens into the existing positional framework by reducing the rotation frequency. This expanded context allowed the model to process multiple cells in sequence, enabling temporal reasoning across a trajectory rather than reasoning about one cell at a time. Stage 2 training used Genecorpus-Aging-22M: approximately 22 million single-cell transcriptomes across roughly 600 human cell types from about 3,800 donors representing every decade of life from birth to 90-plus years, balanced by gender (49% male, 51% female), generating approximately 650 billion tokens. Combined across both stages, MaxToki trained on nearly 1 trillion gene tokens in total.
The Temporal Prompting Strategy
The most architecturally novel contribution of MaxToki is its prompting strategy. A prompt consists of a context trajectory — two or three cell states plus the timelapses between them — followed by a query. The model then performs one of two tasks:
Task 1: Given a context trajectory and a query cell, predict the timelapse (in months) needed to reach that query cell from the last context cell.
Task 2: Given a context trajectory and a query timelapse, generate the transcriptome of the cell that would arise after that duration.
For Task 1, a standard cross-entropy loss is insufficient because it treats each timelapse value as a disconnected category. Instead, the research team used continuous numerical tokenization with a mean-squared error (MSE) loss function, teaching the model that timelapses fall along a numerical continuum. This design choice produced dramatically lower prediction errors — the median prediction error for held-out ages dropped to 87 months with MaxToki, compared to 178 months for a linear SGDRegressor baseline and 180 months for the naive baseline of assuming each query cell was the most common age for that cell type and gender.
Crucially, the model is never explicitly told which cell type or gender it’s dealing with. It infers the trajectory context from the cells themselves — a form of in-context learning. This is why the model generalizes to held-out cell types it never saw during training: it achieves a Pearson correlation of 0.85 between predicted and ground truth timelapses on completely unseen cell type trajectories, and a Pearson correlation of 0.77 on held-out ages from held-out donors.
GPU Engineering at Scale
Training nearly 1 trillion gene tokens required serious infrastructure work. For the 1 billion parameter variant, the team implemented FlashAttention-2 via the NVIDIA BioNeMo stack built on NeMo, Megatron-LM, and Transformer Engine. To enable FlashAttention-2, they modified feed-forward hidden dimensions to be evenly divisible by the number of attention heads — a hard compatibility requirement. Combined with mixed-precision training using bf16, these changes yielded approximately a 5x improvement in training throughput and a 4x increase in achievable micro-batch size on H100 80GB GPUs. For inference, adopting the Megatron-Core DynamicInferenceContext abstraction with key-value caching resulted in over 400x faster autoregressive generation compared to the naive baseline.
What the Model Learned — Without Being Told
Interpretability analysis on the 217 million parameter variant revealed something striking: approximately half of the attention heads learned, entirely through self-supervised training with no gene function labels, to pay significantly higher attention to transcription factors compared to other genes. Transcription factors are master regulators of cell state transitions, but the model discovered their importance on its own.
Ablation studies confirmed that both the context cells and the query cell are equally necessary for accurate predictions — masking either component significantly and equivalently degraded performance. Shuffling genes within the rank value encoding to produce “bag of genes” cells (preserving which genes are present but destroying their relative ordering) also significantly damaged predictions, demonstrating that the model learned to use the relative expression ordering of genes, not merely their presence or absence. Further attention analysis showed that individual heads specialized for different components of the prompt — some attending primarily to context cells, others to timelapse tokens, others to the query — with many heads exhibiting cell type-specific activation patterns across the roughly 60 cell types tested.
One failure mode of generative models is learning to output averaged representations. The research team trained a doublet detector — a classifier distinguishing individual cells from simulated doublets formed by merging two cells of the same cell type — on ground truth cells, then applied it to MaxToki-generated cells. Approximately 95% of generated cells were classified as singlets, confirming that the model produces single-cell resolution transcriptomes rather than blended averages.
Inferring Age Acceleration in Disease — Including Diseases Never Seen During Training
Given the model was trained only on healthy control donors, the research team tested whether it could infer aging signatures in disease states entirely absent from training. The approach: provide a context trajectory of normal cells, then query with a disease cell and test whether the model infers more or less elapsed time compared to an age-matched control cell.
In lung mucosal epithelial cells from donors exposed to heavy smoking, the model inferred approximately 5 years of age acceleration compared to age-matched non-smoking controls — consistent with prior reports linking smoking status to telomere shortening and lung aging signatures. In lung fibroblasts from patients with pulmonary fibrosis — a disease characterized by telomere attrition and cellular senescence — the model inferred approximately 15 years of age acceleration.
The Alzheimer’s disease analysis produced several clinically important findings. In microglia from Alzheimer’s patients drawn from the Mount Sinai NIH Neurobiobank, the model inferred approximately 3 years of age acceleration compared to age-matched controls. This result was replicated in an independent cohort from Duke and Johns Hopkins Alzheimer Disease Research Centers using homeostatic microglia specifically. Critically, this second cohort also included patients with mild cognitive impairment and Alzheimer-resilient patients — individuals who share the same neuropathological changes as Alzheimer’s patients but exhibit no cognitive impairment. The model did not infer age acceleration in homeostatic microglia from either the mild cognitive impairment or resilient groups compared to controls, suggesting these patients may be protected from the disease-related age acceleration in this microglial subtype. This distinction between full Alzheimer’s disease and Alzheimer resilience — captured without any disease-specific training — is one of the most clinically significant findings in the paper.
Conclusion
MaxToki represents a meaningful step forward in how AI models can reason about biological time. By moving beyond single-cell snapshots to model entire trajectories of gene network change across the human lifespan, it addresses a limitation that has constrained computational biology for years. The combination of rank value encoding, continuous numerical tokenization, RoPE-based context extension, and in-context learning allowed the model to generalize to unseen cell types, unseen ages, and even disease states it was never trained on — all while learning, without any supervision, to pay higher attention to the transcription factors that actually drive cell state transitions.
What makes MaxToki particularly compelling for both researchers and engineers is that its predictions did not stop at the computational level. The model nominated novel pro-aging drivers in cardiac cell types that were subsequently validated to cause age-related gene network dysregulation in iPSC-derived cardiomyocytes and measurable cardiac dysfunction in living mice within six weeks — a direct line from in silico screening to in vivo consequence. With pretrained models and training code publicly available, MaxToki offers a reusable framework that the broader community can build on, fine-tune for specific disease contexts, and extend to new tissue types. As longitudinal single-cell datasets continue to grow, temporal foundation models like MaxToki may become a standard tool for identifying intervention points before age-related diseases take hold.
Check out the Paper, Model and Repo. Also, feel free to follow us on Twitter and don’t forget to join our 120k+ ML SubReddit and Subscribe to our Newsletter. Wait! are you on telegram? now you can join us on telegram as well.
Need to partner with us for promoting your GitHub Repo OR Hugging Face Page OR Product Release OR Webinar etc.? Connect with us